Scoring Functions
The fitness of a sequence is determined through a linear combination of metrics, which are computed by various scoring functions. Each scoring function yields two numerical values per evaluation: a metric and a score. The metric serves as a reference value that illustrates the sequence’s optimality according to a particular characteristic. It is primarily designed for human interpretation and appears in reports and logs. On the other hand, the score forms a component of the fitness function, where a higher number corresponds to greater optimality. The balance between the scoring functions is tuned through their weights, which can be adjusted using command line options.
VaxPress comes with a variety of integrated scoring functions. These functions enable optimization with considerations of RNA stability, translation efficiency, and ease of development and manufacturing. VaxPress also provides an add-on system to create and use your scoring functions. This allows you to drive the sequence to include any features you desire.
The following sections will explain the rationale and context behind these built-in scoring functions.
RNA Folding
RNA folding has been shown to be an overwhelming factor determining an mRNA vaccine’s efficacy[1],[2],[3]. The secondary structure influences numerous biochemical and biological properties of mRNA. The base-pairing status confines the susceptibility to ribonucleases or divalent metal ions, which can degrade the RNA. Additionally, local structures near the start codon influence the efficiency of translation initiation.
Predicting this secondary structure, however, is a computationally intensive task due to its cubic complexity. VaxPress is designed to handle this prediction task efficiently by utilizing all available CPU cores. Following the prediction, individual score functions are executed to evaluate the optimality of the predicted structures for use as a vaccine sequence. To keep the computational load manageable, only the minimum free energy (MFE) structure is used to score the fitness of sequences among the many possible structures.
VaxPress predicts a candidate sequence’s MFE structure using ViennaRNA
by default. However, users can select LinearFold using the command
line switch “--folding-engine linearfold”. It’s important to
note that LinearFold cannot be used commercially. Nonetheless, it
offers 2X to 4X faster predictions by more approximations.
Minimum Free Energy (MFE)
🔧 Options related to Minimum Free Energy
The Minimum Free Energy (MFE) indicates the thermodynamic stability of an RNA structure. By giving more weight to this measurement, you can drive the sequence optimization to take more stable structures. These stable structures have been shown to degrade slower and produce more antigens in tests[1],[3].
Since longer RNA sequences inherently have a higher likelihood of forming a stronger, lower MFE, structure. To account for this bias, the MFE score is normalized by dividing it by the sequence length, measured in 1kb units.
Loop Length (total unpaired bases)
🔧 Options related to Loop Length
Loops in RNA secondary structures are typically their most vulnerable points due to their flexibility and susceptibility to degradation-causing substances. The MFE structure usually has a shorter total loop length than most other structures. However, because G:C base pairs are much stronger than A:U pairs, minimizing the total length of loops does not always correlate with the MFE.
Prioritizing loop length in optimization can reduce these vulnerable sections rather than achieve overall thermodynamic stability. As with the MFE score, the total loop length is adjusted by dividing it by the RNA sequence length, scaled in 1kb units.
Start Codon Structure
🔧 Options related to Start Codon Structure
Strong structures near the start codon can disrupt the initiation
of the translation
process[4],[5],[6].
This scoring function aims to reduce the number of base pairings close
to the start codon. You can adjust the width of the leader region
under consideration with the --start-str-width option.
To prioritize this over other factors like MFE and loop length, the
default weight for this function is significantly higher than the
others. However, if the initial sequence has been optimized for
certain features, the high penalty might diminish its existing
optimization. This happens when unfavorable mutations in other
regions are carried along with mutations that reduce the leader
structure penalty. To mitigate this, for the first iterations,
mutations can be restricted to the leader sequence near start codons
using the --conservative-start
option.
Stem Length
🔧 Options related to Stem Length
Long double-stranded RNA regions can trigger a strong immune response as they are detected by the innate immune system’s pattern recognition receptors (PRRs)[7],[8]. This response not only decreases antigen production but also actively degrades mRNAs.
The stem length penalty is based on the total length of stems, without bulges or internal loops, in the MFE structure. By default, the lower limit to be considered “long” is around 30 bp, which includes a safe margin to reduce the chances of appearances of long stems in the non-MFE structures.
The lower threshold, ~10 bp, might also be useful to prevent long inverted repeats, which can hinder efficient chemical synthesis and assembly of gene fragments. Restricting these patterns can improve the productivity of gene synthesis and amplification for mRNA vaccine development and manufacturing.

Codon Usage
Codon usage bias refers to the discrepancies in the frequency of synonymous codons within a coding sequence. This bias can significantly influence the stability of mRNA in cells and the quantity of protein produced[9],[10]. In the case of mRNA vaccines, where antigen production takes place within the cells of the human recipient, it is generally a safe strategy to align the codon frequencies with those found in the human transcriptome.
Codon Adaptation Index (CAI)
🔧 Options related to Codon Adaptation Index
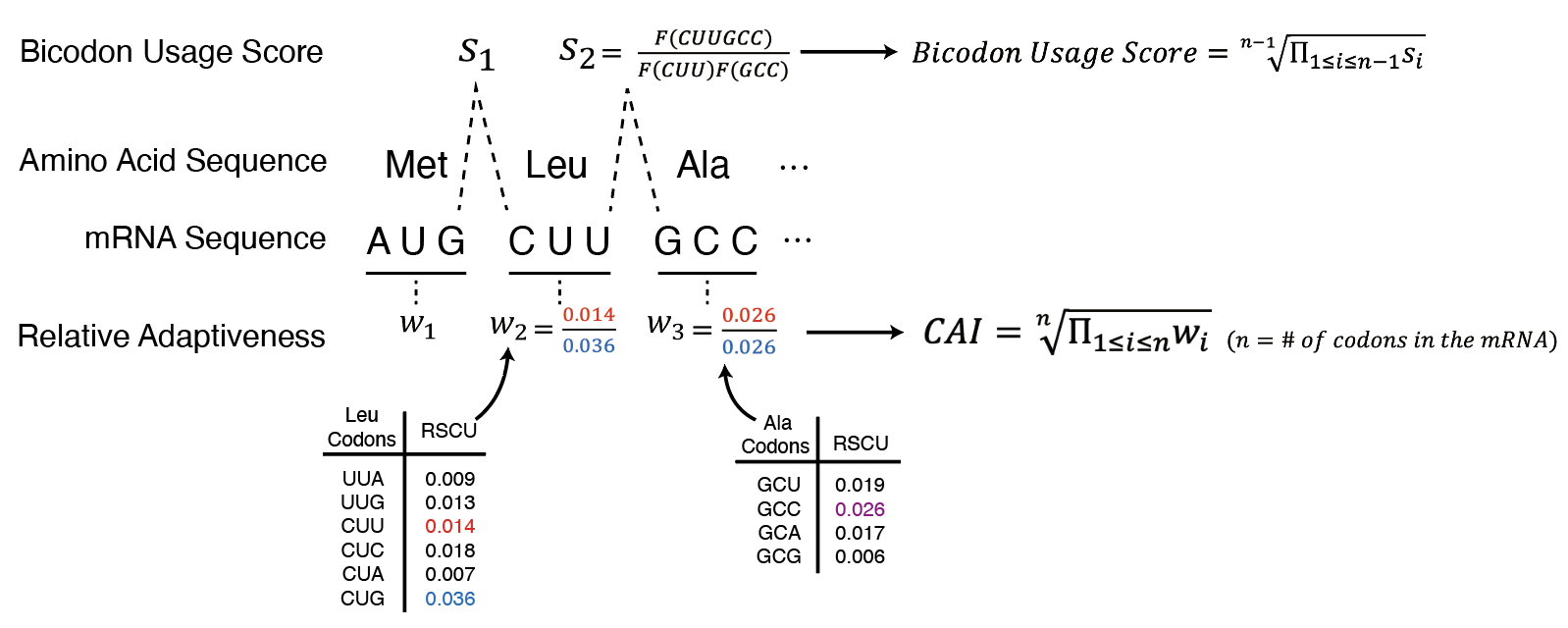
The Codon Adaptation Index (CAI) measures the similarity between the codon usage in a given sequence and a reference sequence[9]. The relative adaptiveness of each codon is calculated based on the the frequency of a specific codon to the most frequently used synonymous codon in highly expressed genes. The score for each codon, \(w_{i}\), is defined as follows:
where \(f_{i}\) is the number of observations for codon \(i\) in the reference sequence, and \(\max{f_{i}}\) is the number of observations for the most frequently used synonymous codon in the reference sequence. The CAI is then calculated as the geometric mean of the relative adaptiveness of all codons in the sequence.
where \(n\) is the number of codons in the sequence.
Bicodon Usage
🔧 Options related to Bicodon Usage
In addition to the biased use of individual codons, the frequency of consecutive codon occurrences is also known to be significantly biased in highly expressed genes in all three kingdoms of life[11]. To account for this, the bicodon usage score is designed to match the codon pair frequencies in the transcriptome of the target species The score is calculated as follows:
where \(ABC,DEF\) represents a codon pair, while \(F(ABC)\) denotes the frequency of the individual codon \(ABC\) within the reference sequence.
The bicodon adaptation score, \(w_{ABC,DEF}\), is standardized to fit within a specific range for easier interpretation before being integrated into the final sequence score.
The score is calculated using a table prepared from the CoCoPUTs codon usage database[12].
Other Activity Factors
Besides RNA secondary structure and codon usage bias, various other sequence properties can influence the overall efficacy of an mRNA vaccine. These properties impact the stability of the vaccine both in solution and within cells, as well as its exposure to interferon responses. The functions summarized in this section aim to enhance the anticipated effectiveness of mRNA vaccines by optimizing those predictive factors.
DegScore
DegScore, a ridge regression model, predicts the degradation rate of each base position in solution, reflecting both primary and secondary structures[3]. It was trained using high-throughput degradation profiles from hundreds of mRNA sequences under moderate and harsh degradation conditions. In VaxPress, the reported metric is the average DegScore across all positions.
iCodon-Predicted Stability
🔧 Options related to iCodon-Predicted Stability
iCodon is a codon optimization tool that enhances the predicted in-cell stability, drawing from high-throughput half-life measurements of endogenous mRNAs[13]. The prediction model primarily relies on the rare codon usage, contingent on the total CDS length. VaxPress integrates iCodon’s in-cell stability prediction as a scoring function, in addition to other optimization factors. iCodon’s current human prediction model is based on SLAM-seq data obtained from K562 cells[14].
Uridine Count
🔧 Options related to Uridine Count
N1-methylpseudouridine is a modification universally employed in the initial FDA-approved mRNA vaccines that effectively addressed the SARS-CoV-2 pandemic. This modification, which substitutes uridines with N1-methylpseudouridine, dramatically improves mRNA vaccines’ translational efficiency and stability[15]. It is thought to alter the action of some pattern recognition receptors or interferon response factors to the mRNA.
Interestingly, depleting uridine improves the translational capacity and decreases excessive immunogenicity for mRNAs, irrespective of the uridine modification to N1-methylpseudouridine[16]. VaxPress tries to reduce the presence of uridines by substituting them with synonymous codons that contain fewer uridines.
Similarly, VaxPress offers an alternative feature that specifically reduces the number of uridines in loops rather than their total count. This function is based on the observation that uridines in loops are the most vulnerable to degradation[3]. For more details, please refer to the relevant section.
Production Factors
Working with a codon-optimized CDS necessitates gene synthesis, typically achieved through chemical oligonucleotide synthesis and in vitro assembly. However, factors like repeats and GC content can complicate this procedure, increasing the likelihood of errors and reducing productivity. The scoring functions described in this section mitigate the potential issues by penalizing the problematic sequences.
Local GC Content
🔧 Options related to Local GC Ratio
Steps in gene synthesis often encounter issues with GC-rich regions. These complications can interfere with the synthesis and amplification of high-GC sections, causing researchers to spend additional time troubleshooting, subsequently extending the overall development period. To mitigate these challenges, VaxPress calculates the GC content within specified intervals and widths of the sequence. The metric is determined from the sum of each bin’s score, with the highest score assigned to a GC ratio of 50% and lower scores given to more biased content. The plot below illustrates the partial score function calculated for each bin.

Repeat Length
🔧 Options related to Repeat Length
The presence of tandem repeats or inverted repeats interferes with
virtually every step in gene synthesis, cloning, and manipulation.
The repeat length function penalizes detected tandem repeats to
minimize the appearance of such repeats in the sequence. Currently,
VaxPress utilizes pytrf’s
GTRFinder to detect tandem repeats. The metric is determined
by the total length of all tandem repeats detected by GTRFinder
that surpass a specified threshold. This approach ensures a more
streamlined and error-free process in the development and manufacturing
of vaccines.